The Power of Bioinformatics Algorithms: Unraveling Evolutionary Relationships with Phylogenetic Trees
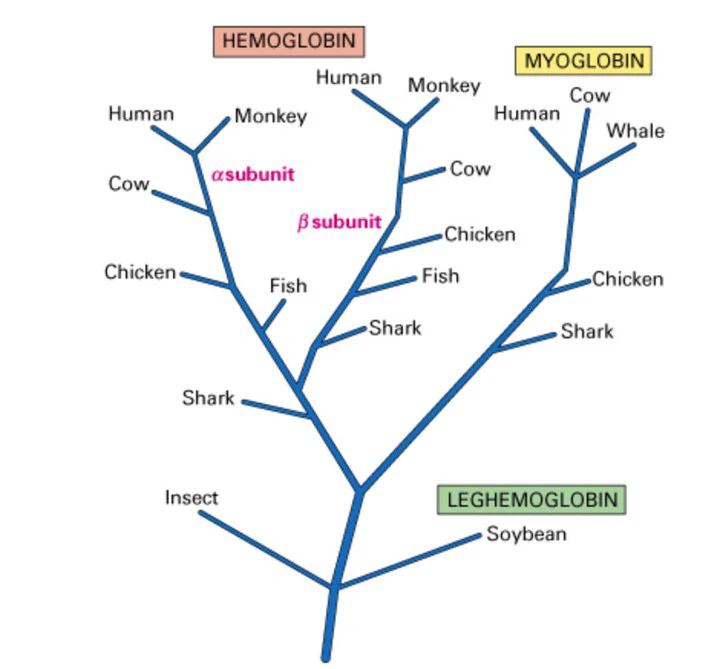 A visual representation of a phylogenetic tree
A visual representation of a phylogenetic treeIntroduction
In the vast and complex world of biological data, understanding the intricate relationships between species and genes is paramount. This is where bioinformatics algorithms, particularly those focused on phylogenetic tree reconstruction, play a pivotal role. As explored in my own thesis, these computational tools are not just academic exercises; they are essential for unlocking profound insights into evolution, gene function, and disease.
You can read my thesis on phylogenetic tree reconstruction and the maximum greedy consensus tree problem Read My Thesis.
What are Phylogenetic Trees?
Phylogenetic trees are branching diagrams that illustrate the evolutionary relationships among various biological entities, such as species, genes, or other taxa. They are hypotheses about the evolutionary history of a set of organisms or genes, showing how they are descended from a common ancestor.
Key concepts in phylogenetics include:
- Species Trees: Represent the evolutionary history of a group of species.
- Gene Trees: Represent the evolutionary history of a particular gene. Gene trees can sometimes differ from species trees due to phenomena like incomplete lineage sorting or gene duplication.
- Gene Duplication: A major evolutionary event where a segment of DNA is duplicated, leading to two copies of a gene within the same genome. Understanding gene duplication events is crucial for inferring accurate evolutionary histories and gene functions.
Real-World Applications
The applications of phylogenetic tree algorithms are incredibly diverse and impactful:
- Evolutionary Biology: Constructing species trees to understand the diversification of life on Earth, inferring ancestral traits, and studying co-evolutionary processes.
- Genomics and Comparative Genomics: Identifying homologous genes across different species, understanding gene family evolution, and detecting gene duplication and loss events.
- Epidemiology: Tracing the spread of viruses and bacteria by constructing phylogenetic trees of pathogens, which aids in understanding outbreaks and developing control strategies.
- Drug Discovery: Identifying conserved protein families and their evolutionary relationships can help in targeting specific proteins for drug development.
- Conservation Biology: Assessing biodiversity and identifying evolutionarily distinct species for conservation efforts.
- Forensics: Using phylogenetic analysis to trace the origin and spread of biological samples.
Connection to My Thesis
My thesis focused on the challenging problem of phylogenetic tree reconstruction and, more specifically, on optimizing the maximum greedy consensus tree problem. This is an NP-hard problem, meaning that finding the absolute optimal solution becomes computationally intractable as the number of input trees grows. My research explored novel algorithmic approaches to efficiently approximate or find optimal solutions for this complex problem, which is crucial for accurately representing evolutionary relationships from diverse biological datasets. This work, including a paper presented at the 12th ICECE, highlighted the continuous innovation required to keep pace with the exponential growth of biological information. You can read my thesis here.
Conclusion
Phylogenetic tree algorithms are fundamental tools in modern biology. They provide the computational backbone for countless discoveries, transforming raw genetic data into actionable biological knowledge about evolutionary relationships, gene duplication, and the history of life. As the field of bioinformatics continues to evolve, these algorithms, and the innovative approaches to optimize them, will remain at the forefront of our ability to understand life itself.